Alzheimer’s is often called “diabetes type 3” because there appears to be a correlation between the two disorders.
After first identifying amyloid β plaques and tau tangles in the brain of a deceased patient in the early 1900’s, Alois Alzheimer was careful to not infer that these structures had caused the dementia in the patient. Until recently, most of the research (at least by drug companies) has been aimed at finding ways to reverse the accumulation, or at least stop the spread of these structures. While drugs have been developed that successfully accomplish that task, it appears that Dr. Alzheimer was right - even when plaques and tangles are decreased in Alzheimer’s patients, cognitive decline continues.
Such distinctive structures seem like such a good place to start when addressing a cure to the disease, so where to next? Recent research has begun exploring other avenues including comorbidities to Alzheimer’s disease. This has proved much more successful, particularly with regards to Type 2 Diabetes (Janson et al. 2004 reported that 81% of Alzheimer’s patients have either Type 2 Diabetes or “Impaired Fasting Glucose”). Treating Alzeheimer's disease patients with diabetes drugs has proved successful, so clearly there is a link between Alzheimer's and insulin. One current hypothesis is that insulin acts as a growth factor in the brain — Talbot et al. 2012 found that even in non-diabetic Alzheimer's patients, insulin was no longer effective as a growth factor. Furthermore, the use of diabetes drugs has also shown to be successful in treating other neurodegenerative conditions such as Parkinson’s Disease and even mTBI/concussions (reviewed in Hölscher 2018). The strong link between Alzheimer's disease and insulin sensitivity has led many researchers to refer to the disease as “diabetes Type 3”.
What we learned about the role of insulin in brain health with the Brain Gauge
The pancreas is not the only place that produces insulin; erratic productivity of insulin in the brain could play a leading role in many disorders. Most textbooks and online references will point you to the pancreas as the body’s site of insulin production. Increasing glucose in the system will trigger the pancreas to produce/release insulin into the blood stream. That has been the standard doctrine for decades. However, in 2011, a paper reported that neurogliaform (NGF) cells produce insulin in the brain (Molnár 2014). Many subsequent papers have confirmed this, and they often point to the brain as playing a role in insulin regulation. Insulin levels can be found to be at least 10-100 times higher in the brain than in the bloodstream. This in itself is pretty good evidence that the brain has different needs from the rest of the body, and that it has a mind of its own when it comes to regulating what it needs (pun intended).
What does this have to do with the Brain Gauge? At the time of the discovery of the NGF cell’s insulin producing capability, our lab had been involved in research in several different neurological disorders that yielded some interesting results. Namely, some individuals with chronic pain, autism and chronic TBI had the common characteristic of having lower than normal feed-forward inhibition (how that measure is obtained is described here), and we were working on a hypothesis that feed-forward inhibition was modulated by the NGF cells, and that this was GABAb mediated (as opposed to GABAa; GABAa vs. GABAb will be discussed in another post). Our finding that feed-forward inhibition was lower than normal in these populations was consistent with the idea that the NGF cells were hypo-active. Could we test a population in which NGF cells were hyper-active? If we could do that, then we could validate that this particular measure of feed-forward inhibition was sensitive to NGF cell activity. If insulin production is low in diabetics, then it was a good bet that the brain’s insulin production would be higher than normal. This idea is simply based on the brain’s ability to take care of itself, such as it does with the diving reflex (other parts of the body shut down when oxygen is low in order to maintain brain function). Increasing activity in the NGF cells would have the effect of increasing insulin in the brain to counter-act the reduced levels of insulin in the blood stream, but increasing the activity of the NGF cell would also increase feedforward inhibition. The results from the study of early stage diabetics (who did not have peripheral neuropathy) was that feedforward inhibition was much greater than normal (as opposed to the other neurological disorders we had studied that were lower than normal – see graph). There are many cognitive deficits that have been documented to be co-morbid with diabetes type 2, and above normal feed-forward inhibition is consistent with that.
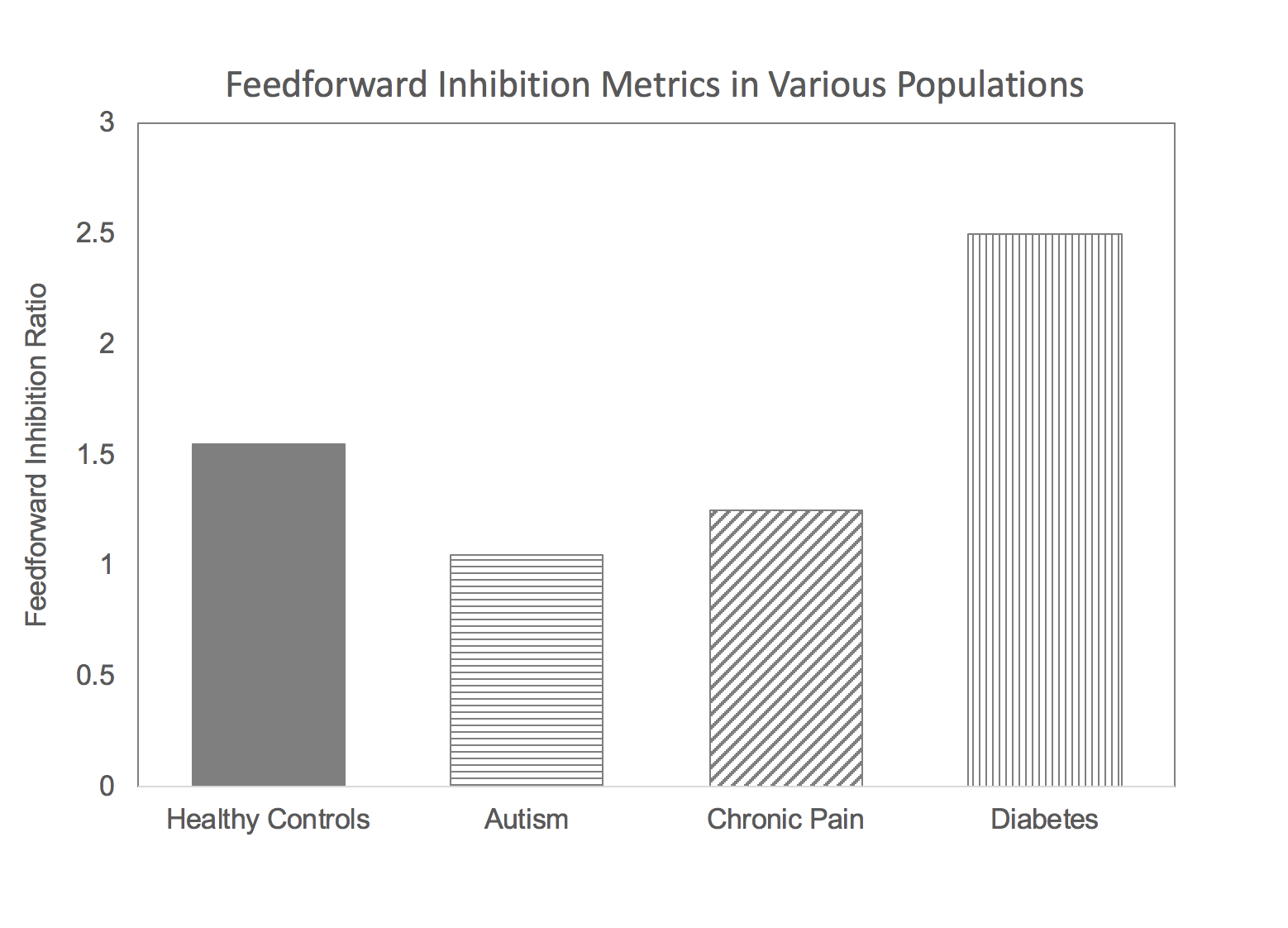
The above graph displays feed-forward inhibition metrics in healthy controls, autism, chronic pain and diabetes populations. Note that the autism and chronic pain groups have values below the average normative value and the diabetes group is well above normative value.
Although feed-forward inhibition is a measure that only researchers are currently obtaining with the Brain Gauge, increases and decreases in that metric parallel degradation in other measures that are more commonly collected with the Brain Gauge. For example, the average data for the diabetics that had the higher than normal feed-forward inhibition metric was significantly worse for several scores. Look at the figure below and compare the brain gauge scores of the healthy controls (left) and the early stage diabetics (right).

Current working hypothesis
As mentioned above, there is quite a bit of literature linking insulin deficiencies with Alzheimer’s (it is even referenced by many as “diabetes type 3”) and diabetes type 2 has been linked with Alzheimer’s epidemiologically. Why would not enough insulin contribute to the degenerative process that is observed in AD? If our working hypothesis is correct, then the tendency to have lower than normal insulin in the blood stream causes the NGF cells to become hyper-active, which causes an imbalance in excitation and inhibition. This imbalance – which also could result in a feedback loop from the brain to that would lower pancreatic insulin production. Lower levels of blood insulin would in turn lead to more insulin production in the brain, which would further exacerbate the condition. The hyperactivity in insulin production would lead to hyperactivity in feed-forward inhibition, which would basically disrupt a cascade of events that culminates in degenerative processes in the brain. The diabetes drugs that improve degenerative disorders break the cycle of the out-of-control feedback loop. While this working hypothesis description is pretty over-simplified (lots of details left out!), the big picture appears to hold up in the literature. You can read more about some of the details here.
One question that is worth asking is – does the degenerative process of Alzheimer's disease initiate the degenerative process of diabetes, does diabetes lead to Alzheimer's disease, or is it a little bit of both? In our opinion, all three options are viable, but it depends on the initial conditions of physical and mental health, environment and genetics. We will continue to investigate and keep you up to date in future posts.
References
Janson J, Laedtke T, Parisi JE, O’Brien P, Petersen RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes. 2004 Feb 1;53(2):474-81.
Hölscher C. Novel dual GLP-1/GIP receptor agonists show neuroprotective effects in Alzheimer's and Parkinson's disease models. Neuropharmacology. 2018 Jan 31.
Molnár G, Faragó N, Kocsis Á et al (2014) GABAergic neurogliaform cells represent local sources of insulin in the cerebral cortex. J Neurosci 34(4):1133-1137.
Talbot K, Wang H-Y, Kazi H, et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. The Journal of Clinical Investigation. 2012;122(4):1316-1338.